 Bioinformatics Recipe Cookbook
Bioinformatics Recipe Cookbook

Results for Bacterial Variant Calling: NZ_CP008918, SRR4124989
Completed
•
Runtime 45 seconds
•
updated 5.4 years ago
by
Istvan Albert
Results generated by running the recipe.
Parameters used during the run:
- Genome Accession Number:NZ_CP008918
- SRA Run Number:SRR4124989
File List
Files created by the recipe run:
plots/counts_by_af.indels.dat
69 bytes
plots/counts_by_af.snps.dat
71 bytes
plots/depth.0.dat
2.5 KB
plots/depth.0.pdf
13.9 KB
 plots/depth.0.png
14.3 KB
plots/depth.0.png
14.3 KB
plots/indels.0.dat
92 bytes
plots/indels.0.pdf
11.0 KB
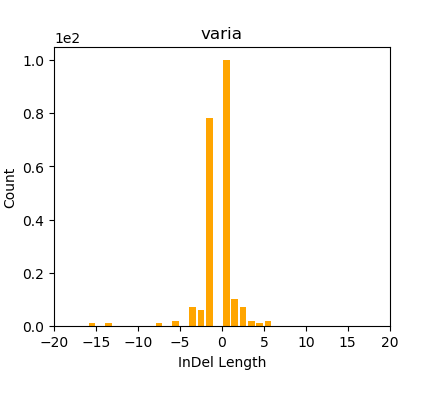 plots/indels.0.png
11.5 KB
plots/indels.0.png
11.5 KB
plots/plot-vcfstats.log
0 bytes
plots/plot.py
8.2 KB
plots/substitutions.0.pdf
10.3 KB
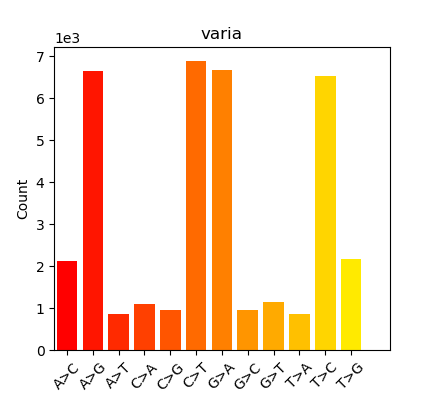 plots/substitutions.0.png
12.7 KB
plots/substitutions.0.png
12.7 KB
plots/tstv_by_af.0.dat
74 bytes
plots/tstv_by_qual.0.dat
88 bytes
reads/SRR4124989_1.fastq
290.8 MB
reads/SRR4124989_2.fastq
290.8 MB
recipe.sh
1.4 KB
ref/NZ_CP008918.fa
2.2 MB
ref/NZ_CP008918.fa.fai
31 bytes
runlog/input.json
1016 bytes
runlog/stderr.txt
1.9 KB
runlog/stdout.txt
369 bytes
variants-stats.txt
9.3 KB
variants.vcf
4.8 MB
Output Messages
Messages printed to the standard output stream:
Read 1047822 spots for SRR4124989 Written 1047822 spots for SRR4124989 *** Sequence statistics file format type num_seqs sum_len min_len avg_len max_len reads/SRR4124989_1.fastq FASTQ DNA 1,047,822 105,830,022 101 101 101 reads/SRR4124989_2.fastq FASTQ DNA 1,047,822 105,830,022 101 101 101 ***
Other Messages
Messages printed to the standard error stream:
+ ACC=NZ_CP008918 + SRR=SRR4124989 + mkdir -p ref + REF=ref/NZ_CP008918.fa + efetch -db nuccore -id NZ_CP008918 -format fasta + mkdir -p reads + fastq-dump --origfmt --split-files -O reads SRR4124989 + echo '*** Sequence statistics' + seqkit stat reads/SRR4124989_1.fastq reads/SRR4124989_2.fastq + echo '*** ' + CPUS=4 + R1=reads/SRR4124989_1.fastq + R2=reads/SRR4124989_2.fastq + samtools sort -l 0 --threads 4 + minimap2 -a -x sr -t 4 ref/NZ_CP008918.fa reads/SRR4124989_1.fastq reads/SRR4124989_2.fastq + bcftools mpileup -Ou -B --min-MQ 60 -f ref/NZ_CP008918.fa - + bcftools call -Ou -v -m - + bcftools norm -Ou -f ref/NZ_CP008918.fa -d all - + bcftools filter -Ov -e 'QUAL<40 || DP<10 || GT!="1/1"' Note: none of --samples-file, --ploidy or --ploidy-file given, assuming all sites are diploid [M::mm_idx_gen::0.048*1.00] collected minimizers [M::mm_idx_gen::0.058*1.46] sorted minimizers [M::main::0.058*1.46] loaded/built the index for 1 target sequence(s) [M::mm_mapopt_update::0.058*1.46] mid_occ = 1000 [M::mm_idx_stat] kmer size: 21; skip: 11; is_hpc: 0; #seq: 1 [M::mm_idx_stat::0.061*1.44] distinct minimizers: 372863 (99.45% are singletons); average occurrences: 1.017; average spacing: 5.993 [M::worker_pipeline::2.762*4.08] mapped 495050 sequences [M::worker_pipeline::4.508*4.22] mapped 495050 sequences [M::worker_pipeline::6.001*4.26] mapped 495050 sequences [M::worker_pipeline::7.181*3.91] mapped 495050 sequences [M::worker_pipeline::7.420*3.80] mapped 115444 sequences [M::main] Version: 2.12-r827 [M::main] CMD: minimap2 -a -x sr -t 4 ref/NZ_CP008918.fa reads/SRR4124989_1.fastq reads/SRR4124989_2.fastq [M::main] Real time: 7.424 sec; CPU: 28.164 sec [bam_sort_core] merging from 0 files and 4 in-memory blocks... [mpileup] 1 samples in 1 input files Lines total/split/realigned/skipped: 45273/0/336/0 + bcftools stats variants.vcf + plot-vcfstats -P -p plots variants-stats.txt Parsing bcftools stats output: variants-stats.txt Plotting graphs: python plot.py
Powered by the
![]() release 2.3.6
release 2.3.6