 Bioinformatics Recipe Cookbook
Bioinformatics Recipe Cookbook
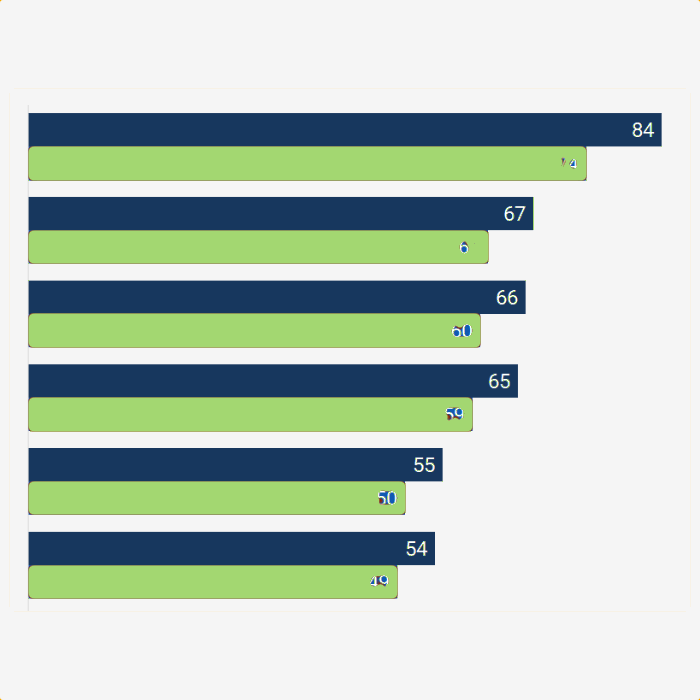
Results for Kraken2 classification validator
Completed
•
Runtime 13 seconds
•
updated 4.1 years ago
by
Istvan Albert
Results generated by running the recipe.
Parameters used during the run:
- Accession numbers:Fish marker accession numbers
File List
Files created by the recipe run:
code/simulate.py
2.3 KB
code/validate.py
3.0 KB
input/accessions.txt
638 bytes
input/reads.fq
13.5 MB
input/reference.fa
807.6 KB
input/taxonomy.txt
5.2 KB
output/kraken2-accuracy.txt
6.1 KB
output/kraken2-output.txt
4.5 MB
output/kraken2-report.txt
14.5 KB
recipe.sh
2.5 KB
runlog/input.json
1.3 KB
runlog/stderr.txt
2.9 KB
runlog/stdout.txt
393 bytes
Output Messages
Messages printed to the standard output stream:
Creating sequence ID to taxonomy ID map (step 1)... Sequence ID to taxonomy ID map already present, skipping map creation. Estimating required capacity (step 2)... Estimated hash table requirement: 1016684 bytes Capacity estimation complete. [0.101s] Building database files (step 3)... Hash table already present, skipping database file build. Database construction complete. [Total: 0.105s]
Other Messages
Messages printed to the standard error stream:
+ set -uex
+ ACC=/export/www/biostar-central/export/media/projects/cookbook/data-275/Fish_marker_sequences
+ N=1000
+ L=100
+ mkdir -p input output code
+ FASTA=input/reference.fa
+ READS=input/reads.fq
+ TAXONOMY=input/taxonomy.txt
+ REPORT=output/kraken2-report.txt
+ OUTPUT=output/kraken2-output.txt
+ ACCURACY=output/kraken2-accuracy.txt
+ cp /export/www/biostar-central/export/media/projects/cookbook/data-275/Fish_marker_sequences input/accessions.txt
+ epost -db nuccore -input /export/www/biostar-central/export/media/projects/cookbook/data-275/Fish_marker_sequences
+ efetch -format fasta
+ epost -db nuccore -input /export/www/biostar-central/export/media/projects/cookbook/data-275/Fish_marker_sequences
+ xtract -pattern DocumentSummary -element Caption,TaxId,Title
+ esummary
++ shasum input/reference.fa
++ cut -c 1-8
+ SHA=cd28e448
+ DB=/home/www/tmp/cd28e448
+ mkdir -p /home/www/tmp/cd28e448
+ ln -sf /home/www/refs/kraken2/taxonomy /home/www/tmp/cd28e448
+ kraken2-build --add-to-library input/reference.fa --db /home/www/tmp/cd28e448
Masking low-complexity regions of new file... done.
Added "input/reference.fa" to library (/home/www/tmp/cd28e448)
+ kraken2-build --build --db /home/www/tmp/cd28e448
+ pip install plac -q
WARNING: pip is being invoked by an old script wrapper. This will fail in a future version of pip.
Please see https://github.com/pypa/pip/issues/5599 for advice on fixing the underlying issue.
To avoid this problem you can invoke Python with '-m pip' instead of running pip directly.
+ URL1=https://raw.githubusercontent.com/biostars/biocode/master/scripts/fasta/simulate.py
+ curl https://raw.githubusercontent.com/biostars/biocode/master/scripts/fasta/simulate.py
% Total % Received % Xferd Average Speed Time Time Time Current
Dload Upload Total Spent Left Speed
0 0 0 0 0 0 0 0 --:--:-- --:--:-- --:--:-- 0
100 2369 100 2369 0 0 11280 0 --:--:-- --:--:-- --:--:-- 11280
+ python code/simulate.py --fname input/reference.fa --count 1000
+ kraken2 -db /home/www/tmp/cd28e448 input/reads.fq --report-zero-counts --report output/kraken2-report.txt
Loading database information... done.
61000 sequences (6.10 Mbp) processed in 0.259s (14108.2 Kseq/m, 1410.82 Mbp/m).
60878 sequences classified (99.80%)
122 sequences unclassified (0.20%)
+ URL2=https://raw.githubusercontent.com/biostars/biocode/master/scripts/classify/validate.py
+ curl https://raw.githubusercontent.com/biostars/biocode/master/scripts/classify/validate.py
% Total % Received % Xferd Average Speed Time Time Time Current
Dload Upload Total Spent Left Speed
0 0 0 0 0 0 0 0 --:--:-- --:--:-- --:--:-- 0
100 3027 100 3027 0 0 13513 0 --:--:-- --:--:-- --:--:-- 13513
+ python code/validate.py -f output/kraken2-output.txt -t input/taxonomy.txt -c 1000
Powered by the
![]() release 2.3.6
release 2.3.6